
Research Interests
The goals of the Computational Structural Biology Lab are to use computational methods to study interesting problems at the interface of biology, physics and chemistry. More generally, the resulting hypothesis or methods are experimentally testable in collaboration with biomedical researchers to gain biological insights. The focus of computational structural biology lab (CSBL) is to have in-depth understanding of autophagy pathway that involves the formation of double-membrane vesicle called autophagosomes. The present-day enormous biological data in hand allow us to investigate various aspects of this pathway in a unique interdisciplinary way. We integrate state-of-art microsecond molecular dynamics simulations contemporary computational data analysis pipeline, and corroborate the models with experime information. Thus, molecular avengers of team CSBL have now the possibility to predict func relevance of proteins by generating physiologically relevant structural models and pinpoint their dis associated properties with unprecedented precision.The broader research themes are mentioned below:
Autophagy
In cell, several biomolecules physically come together to perform a given function. The required coordination for interaction between these molecules such as proteins or lipids constitute the cornerstone of all biological processes. Not surprisingly, abrupt alterations within these complexes result in disease-state of the cell and result in damaging consequences. Atomic modeling and molecular dynamics (MD) simulations provide an opportunity to mimic physiologically-relevant molecular systems and aids in disentangling essential features for a given system under study. Recent advances in technology and novel Al-based tools have significantly improved the gap in timescale and length-scale of biological systems. In particular, the comprehensive understanding of cellular processes at the structural level has remained a formidable challenge until the advent of AlphaFold2 (AF2), an artificial intelligence-based protein structure prediction tool. On the other hand, simulations require high-performance supercomputing infrastructure and the present technological advances have led to an exciting phase in this field, where large supramolecular assemblies are a reality.

SARS CoV-2
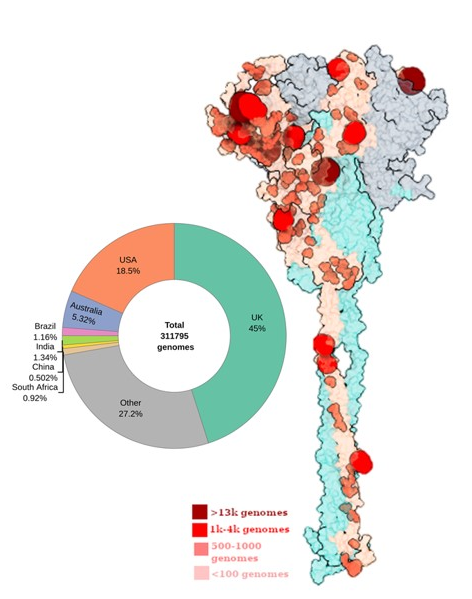
The lab research conducted in the realm of SARS-CoV-2 dynamics has been pivotal in unravelling the intricate molecular mechanisms governing the evolution and behaviour of the SARS-CoV-2 Spike protein. Through detailed analysis of viral genomes and protein structures, our efforts have shed light on the mutational landscape of key viral proteins, particularly the spike protein, across various SARS-CoV-2 variants. In this work, we tracked 527,988 SARS-CoV-2 genomes from publicly available data and identified mutational hotspots based on a temporal acquisition of mutations.By employing advanced computational techniques and molecular dynamics simulations, we've delved deep into understanding the structural and functional implications of critical mutations, such as those found in the Omicron and Delta variants, on viral-host interactions. Our findings have not only elucidated the dynamic nature of viral proteins but also underscored the importance of continuous surveillance and research to combat the ever-evolving threat posed by SARS-CoV-2.
Protein flexibility design and targeting
A fundamental principle in cell biology is the spatial coordination between different cells and within cells that is maintained by compartmentalizing cellular material into membranes. The protein organization within membrane is highly intricate and is tightly linked to location (cell-type specificity) and shape (protein conformational state) that govern a range of signaling activities. Membrane proteins comprise over 39% of human genome. Despite their essential role, they are relatively less studied. More importantly, a large number of the cytoplasmic proteins involved in cell signaling and membrane trafficking reversibly associate with variety of cellular membranes in response to specific stimuli. Membrane recruitment of many such proteins is dependent on chemical modifications, or distinct membrane motifs that impart distinct attributes to protein functionality. The challenge therefore, is to predict mechanism by which cells localize proteins to sites on membrane where they can perform optimally and, in turn, interact with particular substrates. Given the broad spectrum of proteins that interact with membranes, we are set out to understand protein-membrane dynamics in a model pathway - autophagy, a process to degrade cellular waste. In our earlier work, we had deciphered the molecular mechanism of LC3 membrane insertion, a key protein in autophagy. We are further geared up to understand how protein dynamics lead to formation of vesicles called autophagosomes. One of our additional research interests in this theme is to develop methods to aid integration of experimental data with computational representations of biological membranes. We are currently working on development of methods to analyse large-scale structural data of heterogeneous membrane models to infer their mechanistic parameters
AlphaFold2 autophagy pathway
RAPSAP, a curated resource that leverages AI-based AlphaFold2(AF2) and molecular dynamics (MD) simulations to enhance the current structural and functional understanding of autophagic proteins. AF2 enhances the structural space of autophagic proteins by ~47%. RAPSAP enlists the confidence structural predictions of 416 proteins constituting autophagic interactome along with their extensive analysis. The resource also provides comprehensive assessment of 38 core autophagic proteins predicted by AF2. The structures with less template information and high-confidence scores were subjected to microsecond MD simulations to generate ensemble of functionally relevant conformations. The current resource provides an open access to these structural conformers. In addition to the monomeric models, AF2 predicted multimeric complex of ATG7-ATG10 tetramer and its simulated ensemble, is made available. To summarize, RAPSAP serves as an excellent starting point to explore autophagic proteins and complexes in understanding functioning of autophagy pathway

Theurapatics and disease
Autophgy, in general, plays a role in pathological conditions like cancer, especially during the earlier stages of tumorogenesis (4). Different strategies for modulation of autophagy are proposed for tumour selection or degradation of misfolded proteins. Recent developments in autophagy-related drug delivery have revealed several small molecules that target autophagic machinery (ref), in addition, critical protein-protein interaction (PPIs) related to autophagy have also been reported for therapeutical potential (https://www.sciencedirect.com/science/article/pii/S2211383523002769). Therefore, structural information on autophagic proteins is of high importance for the mechanistic and rational drug discovery process. It can also assist in understanding fundamental questions related to the evolution of autophagic proteins, how they interact and also knowledge repositories targeted to autophagy are largely missing.
